Clinical Summary
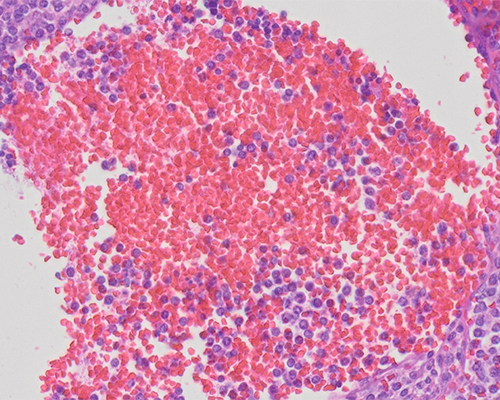
A 62-year-old man presents with sudden onset dizziness. In the emergency department, he appears lethargic and pale. Tachycardia and hypotension are present and physical examination shows moderate abdominal distention, hepatosplenomegaly, and enlargement of groin and cervical lymph nodes. The patient denies fevers, night sweats, or weight loss. Laboratory evaluation shows normocytic anemia with a hemoglobin of 7.2 g/dL and otherwise normal blood cell counts, but rare “atypical” cells are seen on examination of a peripheral blood smear. Emergent laparotomy shows a markedly enlarged, lacerated spleen associated with hemoperitoneum. The splenectomy specimen is a 1,600-gram multinodular spleen. In addition to histologic evaluation, immunohistochemistry is performed and shows the lesional cells to be positive for CD45, CD20, CD79a, CD5, and SOX11.
Master List of Diagnoses
- Diffuse large B-cell lymphoma
- Follicular lymphoma
- Mantle cell lymphoma
- Small lymphocytic lymphoma/chronic lymphocytic leukemia
- Splenic marginal zone lymphoma
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2018, Case 25, and is a mantle cell lymphoma of the spleen.
Criteria for Diagnosis and Comments
This histologic picture, in the context of the immunophenotype, is characteristic of mantle cell lymphoma (MCL). MCL is frequently characterized by a monomorphic population of small to intermediate sized lymphocytes and is typically devoid of centroblasts and paraimmunoblasts. The lymphoid cells in MCL typically have scant cytoplasm and slightly cleaved nuclear contours with indistinct nucleoli. Variant morphologic features include blastic (resembling lymphoblasts), pleomorphic, marginal zone-like (abundant pale cytoplasm), and small cell forms. The characteristic architectural patterns vary from mantle zone to nodular or diffuse patterns.
MCL has a mature B-cell phenotype and, in addition, co-expression of CD5 and CD43 is seen, but the neoplastic cells are characteristically negative for LEF1 (positive in CLL), CD23, BCL6, and CD10. Overexpression of cyclin D1 is a frequent and specific marker of MCL, which itself is the result of a t(11;14) translocation. In the rare cyclin D1 negative MCLs, the transcription factor SOX11 can be used to support the diagnosis. Cyclin D1 and SOX11 are so specific for MCL that, if both are negative, other small B-cell lymphomas must be strongly considered. MCL likely originates from different subtypes of B-cells that express CD5, deriving from pre-germinal center mantle cells (naïve-most common) and post-germinal center mantle cells (somatically mutated-rare).
MCL is a biologically moderately to very aggressive lymphoma with only a small minority of patients being cured or even achieving long-term survival. The majority of patients present with advanced stage disease and have a relatively poor median overall survival (typically 3 - 5 years). Generalized lymphadenopathy and bone marrow involvement are particularly common and seen in a majority of patients. About half of the patients present with involvement of the peripheral blood and spleen; however, splenic rupture like that seen in this patient is rare. Approximately 1 in 5 patients has involvement of the liver and gastrointestinal tract (manifest as “lymphomatoid polyposis” syndrome). Despite the widespread disease at presentation, only a minority of MCL patients have B symptoms.
A high proliferative index (greater than 30%) recognized by Ki-67 immunohistochemistry has been associated with a particularly poor prognosis. This Ki-67 proliferative index improves the prediction of the clinically based Mantle Cell Lymphoma International Prognostic Index (MIPI) and has been included with the clinical criteria. This is called the Biological-MIPI, or MIPI(B).
The differential diagnosis of MCL includes diffuse large B cell lymphoma (DLBCL), but the primary differential considerations are the other small B-cell lymphomas including small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL), follicular lymphoma (FL), and splenic marginal-zone lymphoma (SMZL). MCL can typically be separated from these other lymphomas on histomorphologic grounds, but there can be some overlapping features between these entities requiring the use of ancillary studies to resolve the differential diagnosis. An immunohistochemical panel including CD5, cyclin D1, SOX11, CD10, CD23, and BCL6 typically confirms the diagnosis.
SLL/CLL uncommonly may have a large number of cells with irregular nuclei mimicking the cleaved cells of MCL. Additionally, a subset of SLL/CLL cases may show an interfollicular pattern with “naked residual germinal centers” a feature common to MCL. MCL lacks the larger cells with central nucleoli (prolymphocytes and paraimmunoblasts) that are seen as either isolated or small aggregates in SLL/CLL. Both these lymphomas are positive for CD5, but positive immunohistochemical labeling for cyclin D1 and SOX11 is seen only in MCL. CD23 can be used to differentiate SLL/CLL from MCL as it is positive in the former and negative in the latter.
FL and MCL can have a similar nodular architectural pattern, but the composition of the neoplastic nodules differs. The malignant lymphocytes in MCL have less nuclear irregularities than FL and there are no centroblasts. MCL, unlike FL, shows no immunohistochemical expression for CD10 and BCL6.
SMZL cells have relatively abundant clear cytoplasm that can on occasion be seen in MCL. MCL, however, is more monotonous than MZL with no immunoblasts and plasma cells. CD5, cyclin D1, and SOX11 expression confirms the diagnosis of MCL; MZL is negative for these markers.
DLBCL does not typically provide a differential diagnostic challenge given the large size and pleomorphism of the cells in DLBCL. CD5 and cyclin D1 detection typically facilitates the diagnosis of MCL. However, because CD5 and cyclin D1 labeling can be seen in a small subset of DLBCL cases, these markers need to be evaluated with caution. The cyclin D1 positive DLBCL cases do not however carry the t(11;14) translocation and are SOX11 negative.
Supplementary Questions
- Which of the following best describes mantle cell lymphoma (MCL)?
- MCL is characterized by a proliferation of small to intermediate-sized lymphoid cells with irregular nuclei admixed with centroblasts with prominent nucleoli.
- MCL has a mature B-cell phenotype, typically expresses CD10, and has a t(11;14) translocation leading to overexpression of cyclin D-1.
- Occasional cyclin D1–negative MCLs have been recognized; in these cases, positive immunohistochemical labeling for the transcription factor SOX11 is useful in making the diagnosis.
- Since diffuse large B-cell lymphoma typically expresses cyclin D-1, CD5 is a helpful marker to separate it from MCL.
- While not typical, some cases of MCL have aberrant phenotypes and express CD5, CD43, and BCL6.
- Which of the following is true regarding the clinical features of MCL?
- A high proliferative index recognized by Ki67 immunostaining has been associated with a particularly poor prognosis.
- B symptoms are common at the presentation of MCL.
- The clinical behavior of MCL is usually indolent but a minority can have an aggressive clinical course with a median survival of 3 - 5 years.
- The majority of MCL patients present with localized low stage disease; advanced stage disease is uncommon at presentation.
- The most common extra-nodal site of involvement in MCL is the gastrointestinal tract (manifest as “lymphomatoid polyposis” syndrome).
- Which of the following is the most accurate?
- Generalized lymphadenopathy is common at presentation of MCL, but bone marrow and peripheral blood involvement are rare.
- MCL typically possesses such distinctive histomorphology that use of ancillary studies is typically not needed to make the diagnosis.
- Relatively abundant clear cytoplasm in the lesional cells is typical of MCL.
- Splenic involvement by MCL is seen in about half of patients at presentation but splenic rupture is rare.
- The presence of blast-like and pleomorphic cells essentially rules out a diagnosis of MCL.
References
- Campo E. Mantle Cell Lymphoma. In: Jaffe ES, Arber DA, Campo E, Harris NL, Quintanilla-Martinez L, eds. Hematopathology. Second. Cambridge, MA: Elsevier; 2017:397-414.
- Cheah CY, Seymour JF, Wang ML. Mantle Cell Lymphoma. J Clin Oncol. 2016;34(11):1256-1269.
- Strickland AH, Marsden KA, McArdle J, Lowenthal RM. Pathologic Splenic Rupture as the Presentation of Mantle Cell Lymphoma. Leuk Lymphoma. 2001;41(1-2):197-201.
- Vose JM. Mantle cell lymphoma: 2017 update on diagnosis, risk-stratification, and clinical management. Am J Hematol. 2017;92(8):806-813.
Author
H. Parry Dilworth, MD
CAP Surgical Pathology Committee
Hospital Pathology Associates
Minneapolis, MN
Answer Key
- Occasional cyclin D1–negative MCLs have been recognized; in these cases, positive immunohistochemical labeling for the transcription factor SOX11 is useful in making the diagnosis. (c)
- A high proliferative index recognized by Ki67 immunostaining has been associated with a particularly poor prognosis. (a)
- Splenic involvement by MCL is seen in about half of patients at presentation but splenic rupture is rare. (d)