Clinical Summary
The patient is a 47-year-old African American woman, a teacher by occupation, presenting with worsening dyspnea and nonproductive cough. No significant exposure history is present. Pulmonary function tests reveal a restrictive pattern. High-resolution computed tomography imaging reveals upper lobe-predominant fibrosis, traction bronchiectasis, subpleural honeycombing, reticular opacities, and bulky, bilateral lymphadenopathy. A bronchoalveolar lavage shows elevated CD4 to CD8 T-lymphocyte ratio with negative microbiological studies.
The patient is deemed a candidate for lung transplantation, and sections from the explanted lungs are provided for review. Gross examination shows dilated air spaces interspersed with patchy, irregular fibrosis, and enlarged, firm lymph nodes.
Master List of Diagnoses
- Aspiration pneumonia
- Berylliosis
- Chronic hypersensitivity pneumonitis
- Infectious granulomatosis
- Pulmonary sarcoidosis
- Talc granulomatosis
Archive Case and Diagnosis
This case first appeared as Performance Improvement Program in Surgical Pathology (PIP) 2019, Case 36, and is pulmonary sarcoidosis.
Criteria for Diagnosis and Comments
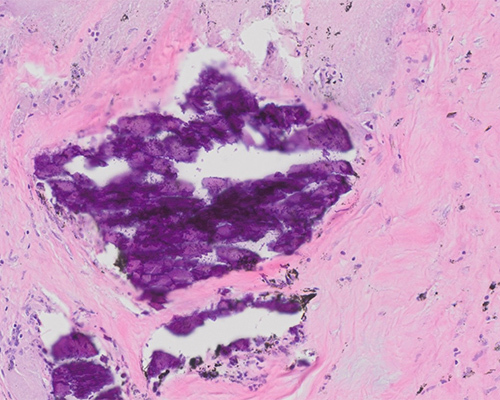
Sections from the lungs show multiple non-necrotizing granulomas distributed along the lymphatics, pleura, and bronchovascular structures. There is a variable amount of microscopic honeycombing and fibrosis. Emphysematous changes are present. Vascular changes of pulmonary hypertension are seen. Lymph nodes show coalescent well-formed granulomas with varying degrees of hyalinization and fibrosis. No polarizable elements are identified in the granulomas. Gomori methenamine-silver (GMS) nitrate stain and acid-fast bacilli (AFB) stains are negative for microorganisms. Clinical and morphological findings are compatible with the diagnosis of pulmonary sarcoidosis.
Sarcoidosis is a multisystem disorder of unknown etiology characterized by non-necrotizing granulomatous inflammation. The fundamental abnormality is altered immune regulation in genetically susceptible individuals. Lungs and bilateral hilar lymph nodes are involved in approximately 90% of patients with CD4+ T cells acting as cardinal players.
Since several infectious and noninfectious etiologies can produce granulomatous inflammation in the lung, the diagnosis of sarcoidosis is always a diagnosis of exclusion. Sarcoidosis affects all races and ethnic groups, though it is particularly common in the Danish, Swedish, and African American population. It is a disease of young individuals between 20 and 50 years of age, with no sex predilection. It is one of the few pulmonary disorders prevalent among nonsmokers.
Clinical presentation of the disease varies according to the site of involvement. The vast majority of patients are asymptomatic, and the diagnosis may incidentally be made based on imaging findings. Symptoms can be nonspecific and include fatigue, low-grade fever, and night sweats, while specific pulmonary symptoms include cough and dyspnea. Pulmonary hypertension is seen in approximately 5%-15% of asymptomatic patients and 50% of symptomatic patients. The incidence of pulmonary hypertension is even higher in patients requiring transplantation. Pulmonary sarcoid is staged by radiographic findings of mediastinal lymph nodes and lung parenchyma (Table 1). Non-pulmonary findings include hilar and mediastinal lymphadenopathy, uveitis, retinitis, cranial nerve palsies, and skin lesions (erythema nodosum and lupus pernio).
Table 1: Clinical Staging of Pulmonary Sarcoidosis by Radiographic Findings
Stage 0 | No mediastinal lymphadenopathy or lung infiltrates |
Stage 1 | Bilateral hilar lymphadenopathy |
Stage 2 | Bilateral hilar lymphadenopathy and reticulonodular infiltrates |
Stage 3 | Bilateral infiltrates only (without lymphadenopathy)* |
Stage 4 | Fibrocystic changes, hilar retraction, cystic and bullous changes |
*In the fibrotic phase of the disease, the lymph nodes are also fibrotic and burnt out with sparse granulomas. Lymph node enlargement may not be present in this phase.
Sarcoidosis is characterized histologically by well-formed, non-necrotizing granulomas that show striking distribution along lymphatics. These granulomas are also distributed along bronchovascular bundles, pleura and interlobular septa. The intervening lung parenchyma is generally spared of significant interstitial inflammation and organizing pneumonia. The granulomas are comprised of epithelioid histiocytes with occasional multinucleated giant cells and a thin rim of sparse lymphocytes (so-called “naked” granulomas). Multinucleated giant cells may show inclusions (asteroid bodies) and calcifications (Schaumann bodies), findings that are not specific to sarcoidosis. In the fibrotic phase, there is hyalinized fibrosis with lack of inflammation. The fibrosis follows the distribution pathway of the granulomas. Eventually, the granulomas may be entirely replaced by fibrosis and only rare, scattered, residual giant cells may be left behind.
The diagnosis of sarcoidosis can be easily made on bronchial biopsy, mediastinal lymph node biopsy, or fine needle aspiration. The yield of finding granulomas in bronchial biopsies is high due to the high incidence of airway involvement. The yield on transbronchial needle aspiration is further increased (to approximately 90%) with the concurrent use of endobronchial ultrasound.
Aspiration pneumonia can be clinically overt or subclinical in nature. Aspiration is associated with acute bronchopneumonia in the early stages and is followed by airspace organization. Foreign body-type granulomas, namely multinucleated giant cells with aspirated foreign material (vegetable/pill matter) may be seen.
Berylliosis is an occupational granulomatous lung disease (pneumoconiosis) caused by exposure to inhalation of dusts containing beryllium or its salts. Acute berylliosis results in diffuse alveolar damage. Chronic forms of berylliosis are virtually indistinguishable from sarcoidosis. The granulomas in chronic berylliosis can be well-formed with large centrally hyalinized nodules, or they can be ill-formed.. Occupational exposure and documented sensitization to beryllium (beryllium lymphocyte proliferation test on blood and/or bronchoalveolar lavage) help clinch the correct diagnosis.
Chronic hypersensitivity pneumonitis (HP) is a chronic inflammatory reaction to inhaled environmental “organic” protein antigens. A whole range of antigens have been implicated; some of the common antigens include moldy hay (farmer’s lung, from exposure to thermophilic actinomycetes in hay) and avian antigens (bird fancier’s lung). Histopathological features include an upper lobe-predominant chronic interstitial pneumonitis, with bronchiolitis and scattered, small, ill-formed, non-necrotizing granulomas in the interstitium Refractile oxalate crystals may be seen within giant cells. Foci of organizing pneumonia may be seen in up to 60% of cases. In the chronic forms of HP, peribronchiolar metaplasia, peribronchiolar fibrosis, and “bridging” fibrosis are frequent findings.
Infectious granulomatosis is the most common cause of pulmonary granuloma. An infectious etiology always needs to be excluded before the consideration of non-infectious granulomatous lesions. The most common organisms found in granulomatous lungs are mycobacteria and fungi. Both necrotizing and non-necrotizing granulomas may be seen in infectious causes. Special stains (GMS, PAS, and AFB) need to be performed on all granulomatous lesions, whether they are necrotizing or non-necrotizing, although the yield is likely to be higher in necrotizing lesions. The yield of identifying an infectious agent is improved by staining multiple tissue blocks. Finally, polymerase chain reaction (PCR) for infectious organisms may be attempted on formalin-fixed paraffin embedded tissues if there is a high index of clinical suspicion.
Talc granulomatosis (talc=hydrated magnesium silicate) can also occur. Talc is used in many industries, and as a “filler” substance for oral medications. Exposure may occur in individuals using an excess of talcum powder. In inhalational talcosis, talc deposits are noted in a perivascular and peribronchial distribution. Talc particles are needle-like, bluish-gray particles that are brightly birefringent under polarized light. In intravenous drug abusers, talc granulomatosis-a reaction caused by a foreign body-type granulomatous response to entrapped talc particles in pulmonary arterioles and capillary beds (ie, alveolar septal distribution) results. The presence of interstitial granulomas with birefringent material raises the possibility of sarcoidosis. The granulomas of sarcoidosis are discrete, well-formed, and round, and they show a characteristic lymphangitic pattern of spread rather than a pure alveolar septal pattern of distribution. Although birefringent material may be seen in sarcoid granulomas, these tend to be sparse when compared to talc granulomatosis.
Supplementary Questions
- Which of the following entities is characterized by chronic interstitial pneumonitis with bronchiolitis,
peribronchial fibrosis, and interstitial ill-formed granulomas?
- Berylliosis
- Chronic hypersensitivity pneumonitis
- Infectious granulomatosis
- Pulmonary sarcoidosis
- Talc granulomatosis
- Which of the following entities is characterized by septal granulomas, vascular thrombi, and foreign body-type
giant cell reaction to needle-like, birefringent particles within giant cells?
- Berylliosis
- Chronic hypersensitivity pneumonitis
- Infectious granulomatosis
- Inhalational talcosis
- Intravenous talcosis
- Pulmonary sarcoidosis
- Which of the following is the typical flow cytometry finding in the bronchoalveolar lavage fluid of a sarcoidosis
patient?
- Clonal B cell population, CD5-, CD10-
- Decreased CD4 to CD8 ratio
- Dim CD5 expression in T-cells
- Elevated CD4 to CD8 ratio
- Elevated CD20 to CD3 ratio
References
- Eishi Y. Etiologic link between sarcoidosis and Propionibacterium acnes. Respir Investig. 2013;51(2):56-68.
- Esteves T, Aparicio G, Garcia-Patos V. Is there any association between sarcoidosis and infectious agents?: a systematic review and meta-analysis. BMC Pulm Med. 2016;16(1):165.
- Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153-2165.
- Kumar V, Abbas A, Aster JC. eds. Robbins Basic Pathology,9th Edition Elsevier Saunders, Philadelphia, PA. 2012. Pages:476-482
- Leslie KO, Wick MR, eds. Practical Pulmonary Pathology: A Diagnostic Approach, 2nd Edition, Elsevier Saunders, Philadelphia, PA, 2011. Chronic Diffuse Lung Diseases: Pages 248-256
- Mukhopadhyay S, Farver CF, Vaszar LT, et al. Causes of pulmonary granulomas: a retrospective study of 500 cases from seven countries. J Clin Pathol. 2012;65(1):51-57.
- Mukhopadhyay S, Gal AA. Granulomatous lung disease: an approach to the differential diagnosis. Arch Pathol Lab Med. 2010;134(5):667-690.
- Mukhopadhyay S. Role of histology in the diagnosis of infectious causes of granulomatous lung disease. Curr Opin Pulm Med. 2011;17(3):189-196.
- Spagnolo P, Rossi G, Trisolini R, Sverzellati N, Baughman RP, Wells AU. Pulmonary sarcoidosis. Lancet Respir Med. 2018;6(5):389-402.
Author
Vijayalakshmi Ananthanarayanan, MBBS, MD, FCAP
Surgical Pathology Committee
Loyola University Medical Center
Maywood, IL
Answer Key
- Chronic hypersensitivity pneumonitis (b)
- Intravenous talcosis (e)
- Elevated CD4 to CD8 ratio (d)

