- Home
- Member Resources
- Articles
- Emerging Family of Spindle Cell Neoplasms with Kinase Fusions
Kinase-rearranged spindle cell tumors have received increasing interest over the past five years, in part due to broader implementation of next-generation sequencing, and the approval of targeted therapies for solid tumors with kinase fusions. While the risk of malignancy and prognosis based on tumor subtype are variable, targeted therapy can be helpful in both the metastatic setting and in patients for whom surgical resection would cause significant morbidity.1,2 Infantile fibrosarcoma (IFS) and inflammatory myofibroblastic tumor (IMT) are the classic spindle cell neoplasms driven by kinase fusions; however, newer entities such as NTRK-rearranged spindle cell tumors and an emerging family of other kinase-driven spindle cell tumors of uncertain biologic potential have recently been recognized (Table 1).
Table 1. Subtypes of Kinase-rearranged Spindle Cell Neoplasms
Tumor type |
Kinase genes involved |
Clinical behavior |
Clinical presentation |
Histologic features |
IHC |
Infantile fibrosarcoma |
NTRK3>>>NTRK1/2, RET, MET, RAF1, BRAF, ALK |
May recur, rarely metastasizing |
Infants; extremities; rapidly growing |
Densely cellular, fascicular, primitive spindle cells |
Pan-TRK+ |
Inflammatory myofibroblastic tumor **Epithelioid inflammatory myofibroblastic sarcoma |
ALK>> ROS1, NTRK3, RET, PDGFRB **RANBP2::ALK, RRBP1::ALK |
May recur, rarely metastasizing **Clinically aggressive |
Children and young adults; visceral and central body cavity sites |
Variable cellularity/histologic features, brisk inflammation |
ALK >> ROS1; SMA, desmin |
Kinase-rearranged spindle cell neoplasms |
NTRK1>>NTRK2/3, RET, RAF1, BRAF, ALK, ROS1, MET |
May recur, rarely metastasizing |
Wide age range; subcutaneous soft tissue or bone |
Lipofibromatosis-like infiltration of fat, keloidal stromal collagen, perivascular hyalinization |
S100, CD34, Pan-TRK >> ALK, ROS1 |
Superficial ALK-rearranged myxoid spindle cell neoplasm |
ALK |
Clinically indolent |
Wide age range; dermal/subcutaneous |
Whorls in myxohyaline stroma |
ALK, S100, CD34 |
Abbreviation: IHC, immunohistochemistry.
Since the FDA approval of larotrectinib, and later entrectinib, for solid tumors harboring NTRK fusions, targeted therapy has been a successful modality of therapy for patients with metastatic or locally aggressive IFS.3 Similarly, therapies such as trametinib (for BRAF and RAF1-rearranged spindle cell neoplasms) and crizotinib (targeting ALK and ROS1-rearranged IMT) have been successfully utilized to improve patient outcomes.4 Recognition of these entities is increasingly important, given the broader range of targeted therapies now available to patients.
Subtypes of kinase fusion-driven spindle cell neoplasms
Infantile fibrosarcoma
IFS, the first malignant spindle cell neoplasm to be characterized by a kinase gene rearrangement, classically harbors t(12;15), leading to ETV6::NTRK3 in up to 90% of cases. IFS presents as a rapidly growing mass in the extremities of infants, congenitally in a third of cases. However, these lesions can also arise in challenging locations to manage surgically, such as the head and neck. While mortality is low and metastatic disease rare, recurrence after excision is not uncommon. IFS is a poorly circumscribed densely cellular spindle cell neoplasm composed of primitive monomorphic spindle cells arranged in tight intersecting fascicles (Figure 1A-1B). Infiltration of fat and muscle fibers is common; some tumors display areas with a myxoid matrix. In the 20 years since its original description, tumors that fit well morphologically and clinically with IFS have now been reported with alternate fusion partners to NTRK3, as well as with other kinase fusions in NTRK1/2, RET, MET, RAF1, BRAF, ALK, and
ABL.5-9
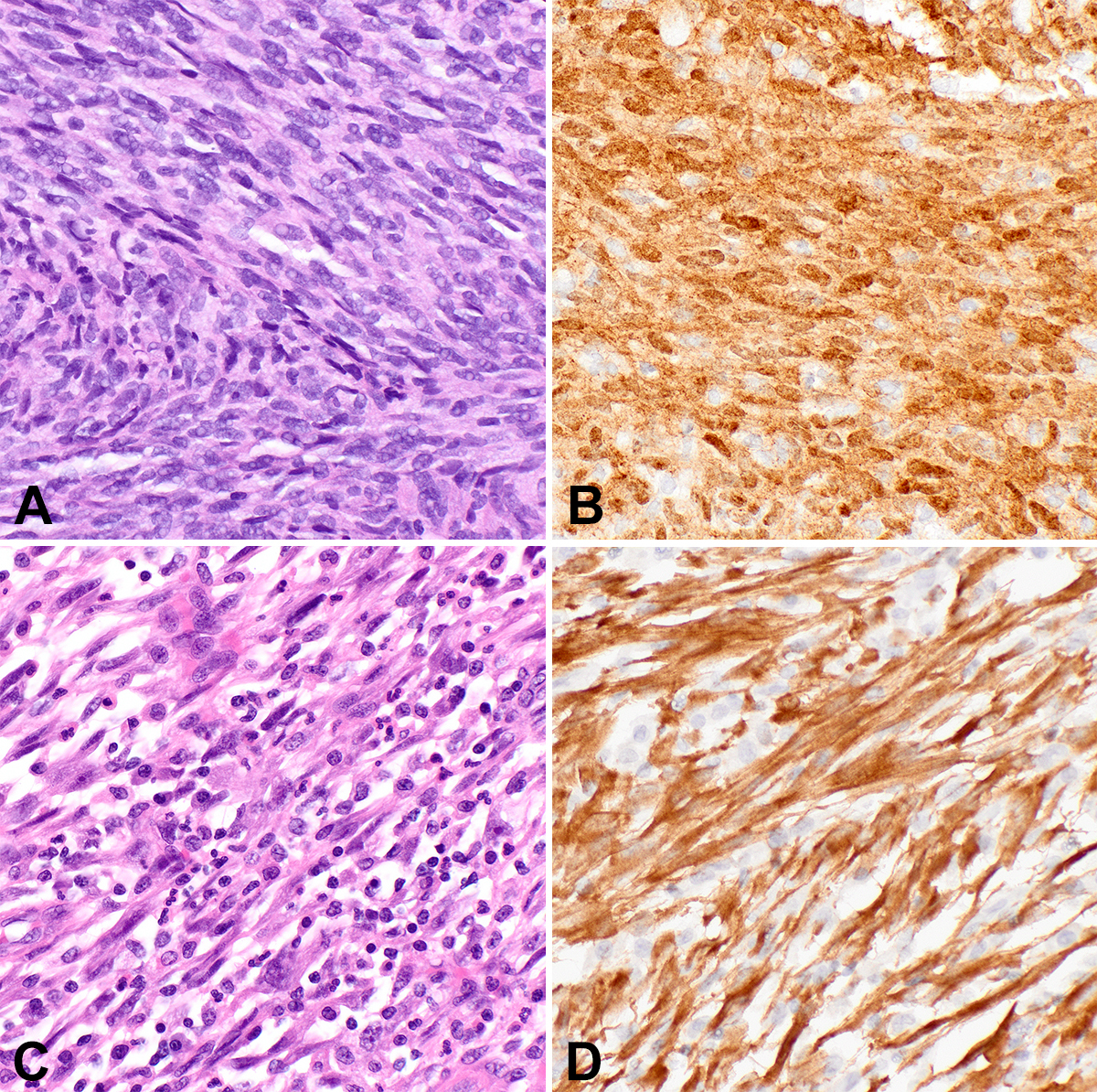
Inflammatory myofibroblastic tumor
IMT, classically harboring ALK gene rearrangements, is a myofibroblastic neoplasm of intermediate biologic potential that arises in children and young adults in visceral and central body cavity locations such as the lung, gastrointestinal tract, and abdomen, but may arise at any anatomic location. IMTs are histologically quite variable, displaying either a fasciitis-like myxoid pattern with a prominent inflammatory infiltrate (Figure 1C), a cellular spindle cell pattern with collagenous stroma and fascicular architecture, or a hypocellular pattern with dense stromal collagen that may mimic desmoid fibromatosis. Immunohistochemistry (IHC) is typically positive for SMA and desmin, with 60% of lesions positive for ALK (Figure 1D). IMTs can recur following excision and rarely metastasize. ALK-negative IMTs have been identified with alternative kinase fusions, including ROS1, NTRK3, RET, and PDGFRB.10,11 A clinically aggressive variant important to recognize is epithelioid inflammatory myofibroblastic sarcoma (EIMS), which presents intra-abdominally, shows epithelioid morphology with prominent nucleoli and a nuclear membrane or perinuclear pattern of ALK staining, and harbors RANBP2::ALK or RRBP1::ALK fusions.12
Kinase-rearranged spindle cell neoplasms
NTRK-rearranged spindle cell tumors that do not clinically or morphologically fit with IFS were first described in 2016 as lipofibromatosis-like neural tumors,13 benign subcutaneous lesions presenting in children and young adults with a wide anatomic distribution. These cellular spindle cell neoplasms show minimal nuclear atypia and a distinct lipofibromatosis-like pattern of infiltration into fat and skeletal muscle. An additional feature in some lesions is thick stromal collagen bundles. By IHC, these tumors are positive for S100 protein, CD34, and Pan-TRK but are negative for SOX10, and harbor NTRK1 rearrangements. Subsequently, NTRK-rearranged neoplasms with other features were described in both children and adults, in bone and soft tissue, broadening the range of morphology and clinical behavior.6,14 Some tumors are characterized by a relatively bland, monomorphic spindle cell appearance with prominent keloidal stromal collagen deposition and perivascular hyalinization (Figure 2). Other lesions are more cellular and fascicular, with increased mitotic activity, and can appear frankly malignant, with aggressive clinical courses including metastases.
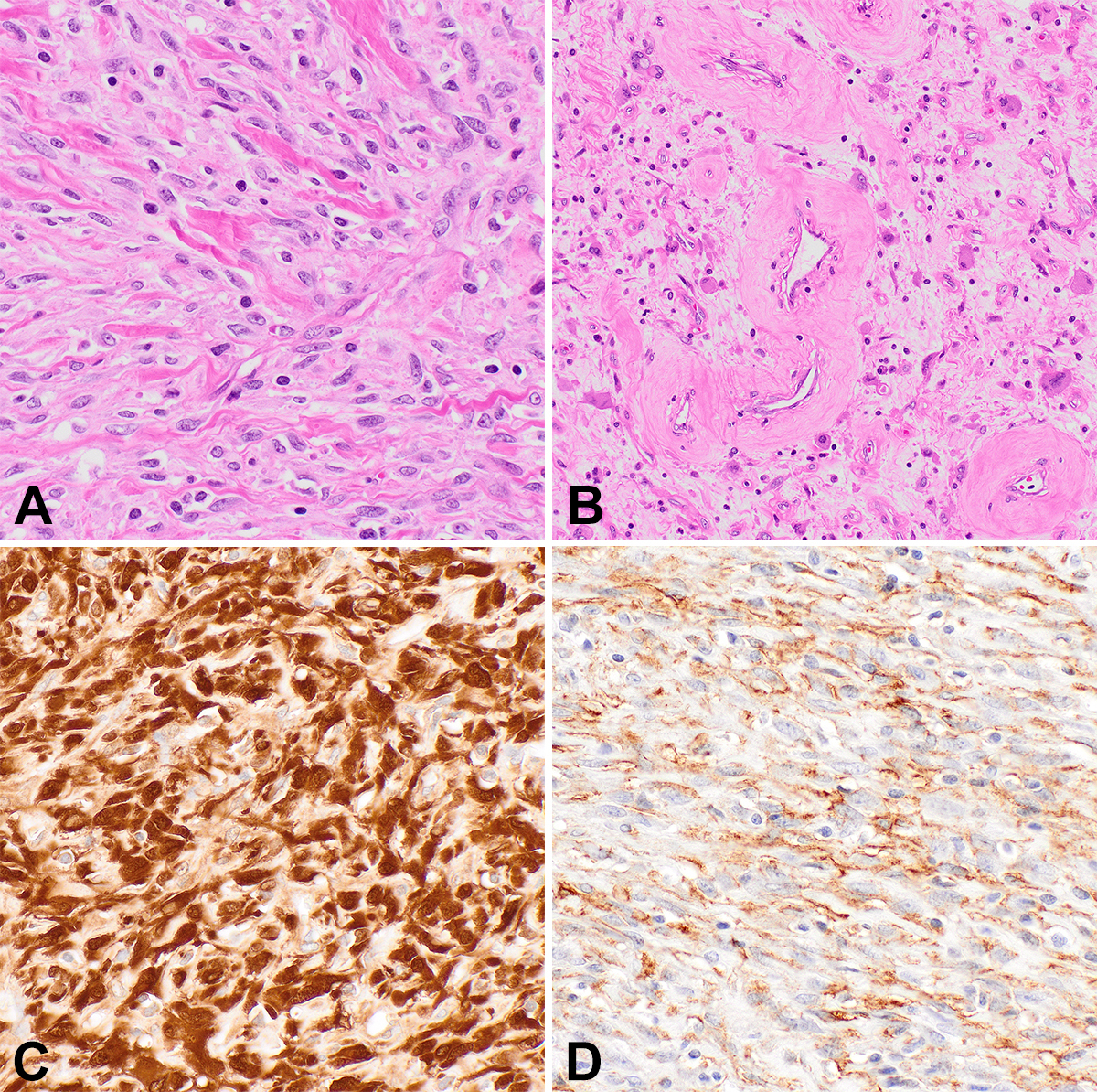
Since the recent descriptions of NTRK-rearranged spindle cell tumors, case reports and series have reported tumors with similar features but rearrangements in other kinase genes, such as RET, RAF1, BRAF, ALK, ROS1, and MET.7,8,14-16 As in NTRK-rearranged spindle cell tumors, perivascular hyalinization and keloidal stromal collagen are common, although such tumors display a wide morphologic spectrum as well (Figure 3). IHC is typically positive for CD34 and/or S100 protein, similar to tumors with NTRK fusions. Although metastases and poor outcomes appear to be uncommon in these tumors, malignant behavior is possible, and tumors may present at surgically challenging anatomic locations; therefore, affected patients may benefit from targeted therapies.
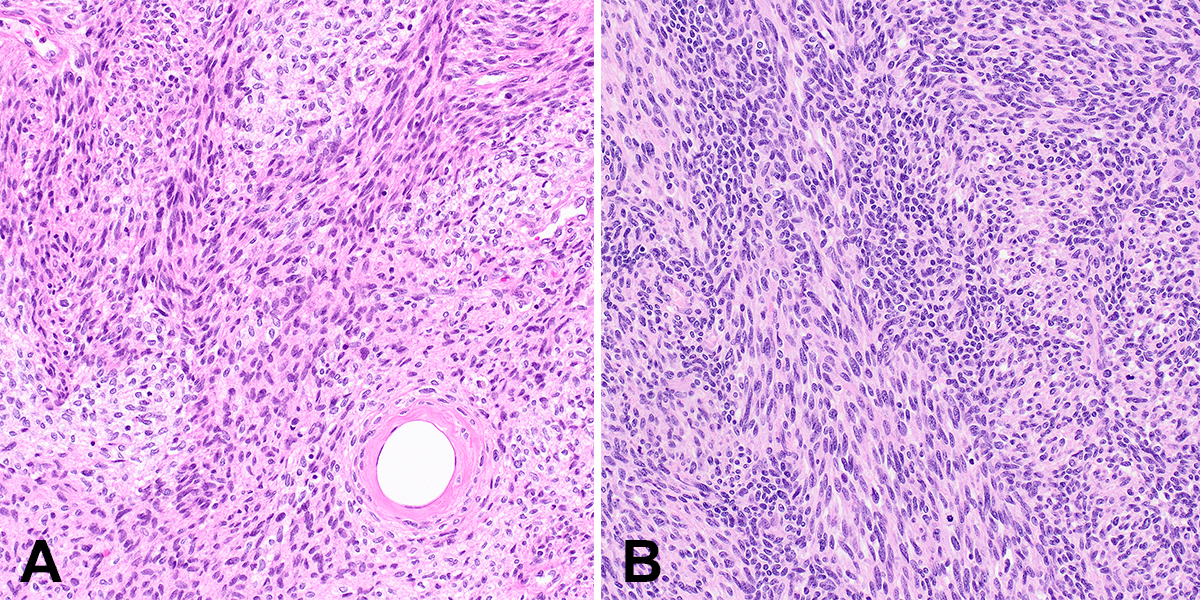
Superficial ALK-rearranged myxoid spindle cell neoplasm
Recently, a novel cutaneous ALK‑rearranged spindle cell tumor was described, provisionally termed superficial ALK-rearranged myxoid spindle cell neoplasm.17 These are superficial lesions involving the dermis and subcutis, which present on the trunk and lower extremities over a wide age range. Histologically, these lesions are composed of bland spindled to ovoid cells in a myxoid to myxohyaline stroma and typically show a biphasic pattern of alternating hypocellular areas and more tightly whorled cellular areas with plump nuclei. IHC is diffusely positive for ALK, CD34, and S100 protein and negative for SMA, EMA, keratins, and SOX10. All reported tumors harbored ALK fusions preserving the ALK kinase domain (breakpoints 5’ to exon 20) with partners previously not reported in IMT, including FLNA, MYH10, and HMBOX1. Although follow up is thus far limited and cases reported in the literature few, recurrences and metastases were not identified, even when excised with positive margins.
Molecular techniques for detecting kinase fusions
Given the central role of molecular diagnostics for classifying these lesions, pathologists should understand the different molecular techniques for detecting a kinase fusion. The classic cytogenetic methodologies of karyotype and fluorescence in situ hybridization (FISH) are options for detecting chromosomal and gene rearrangements, although the former requires fresh tissue for culture, considerable expertise to perform and analyze and may take several weeks. FISH, in contrast, has a rapid turnaround time, is widely available, and is relatively simple to perform, but will not detect the fusion partner or give information about breakpoints. FISH thus requires selection of the gene suspected to be rearranged and is not an efficient modality for lesions where numerous kinase gene fusions are possible. Next generation sequencing via DNA-based hybrid capture or RNA-based anchored multiplex PCR are both expensive assays with slower turnaround times; however, these techniques allow for testing of multiple fusion genes and will show breakpoints and fusion partners. Finally, IHC for ALK, Pan-TRK, and ROS1 are widely available, inexpensive, and rapid, and can be performed as screening (for Pan-TRK and ROS1) or diagnosis (for ALK) in cases where IFS, IMT, or a kinase-rearranged spindle cell tumor is highly suspected.
In summary, histologic recognition of kinase-driven spindle cell tumors is important; confirmation with appropriate molecular diagnostic testing is critical in order for oncologists to select optimal treatment for their patients. While the behavior of kinase-driven spindle cell tumors is highly variable, most with an excellent prognosis, targeted therapy is needed in some cases and has been successfully applied. Kinase-driven spindle cell neoplasms can be challenging to recognize, given their rarity, particularly in small biopsy samples. Therefore, pathologists should have a low threshold to recommend molecular diagnostics to identify a kinase fusion in difficult-to-classify histologically uniform spindle cell neoplasms.
References
1. Butrynski JE, D’Adamo DR, Hornick JL, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363(18):1727–1733. doi:10.1056/NEJMoa1007056
2. Laetsch TW, DuBois SG, Mascarenhas L, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018;19(5):705–714. doi:10.1016/S1470-2045(18)30119-0
3. DuBois SG, Laetsch TW, Federman N, et al. The use of neoadjuvant larotrectinib in the management of children with locally advanced TRK fusion sarcomas. Cancer. 2018;124(21):4241–4247. doi:10.1002/cncr.31701
4. Baranov E, Winsnes K, O’Brien M, et al. Histologic characterization of paediatric mesenchymal neoplasms treated with kinase-targeted therapy. Histopathology. 2022;81(2):215–227. doi:10.1111/his.14680
5. Penning AJ, Al-Ibraheemi A, Michal M, et al. Novel BRAF gene fusions and activating point mutations in spindle cell sarcomas with histologic overlap with infantile fibrosarcoma. Mod Pathol. 2021;34(8):1530–1540. doi:10.1038/s41379-021-00806-w
6. Davis JL, Lockwood CM, Stohr B, et al. Expanding the spectrum of pediatric NTRK-rearranged mesenchymal tumors. Am J Surg Pathol. 2019;43(4):435–445. doi:10.1097/PAS.0000000000001203
7. Davis JL, Vargas SO, Rudzinski ER, et al. Recurrent RET gene fusions in paediatric spindle mesenchymal neoplasms. Histopathology. 2020;76(7):1032–1041. doi:10.1111/his.14082
8. Kao Y-C, Fletcher CDM, Alaggio R, et al. Recurrent BRAF gene fusions in a subset of pediatric spindle cell sarcomas: expanding the genetic spectrum of tumors with overlapping features with infantile fibrosarcoma. Am J Surg Pathol. 2018;42(1):28–38. doi:10.1097/PAS.0000000000000938
9. Choo F, Rakheja D, Davis LE, et al. GAB1-ABL1 fusions in tumors that have histologic overlap with NTRK-rearranged spindle cell tumors. Genes Chromosomes Cancer. 2021;60(9):623–630. doi:10.1002/gcc.22972
10. Antonescu CR, Suurmeijer AJH, Zhang L, et al. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am J Surg Pathol. 2015;39(7):957–967. doi:10.1097/PAS.0000000000000404.
11. Yamamoto H, Yoshida A, Taguchi K, et al. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology. 2016;69(1):72–83. doi:10.1111/his.12910
12. Mariño-Enríquez A, Wang W-L, Roy A, et al. Epithelioid inflammatory myofibroblastic sarcoma: an aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol. 2011;35(1):135–144. doi:10.1097/PAS.0b013e318200cfd5
13. Agaram NP, Zhang L, Sung Y-S, et al. Recurrent NTRK1 gene fusions define a novel subset of locally aggressive lipofibromatosis-like neural tumors. Am J Surg Pathol. 2016;40(10):1407–1416. doi:10.1097/PAS.0000000000000675
14. Suurmeijer AJH, Dickson BC, Swanson D, et al. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer. 2018;57(12):611–621. doi:10.1002/gcc.22671
15. Antonescu CR, Dickson BC, Swanson D, et al. Spindle cell tumors with RET gene fusions exhibit a morphologic spectrum akin to tumors with NTRK gene fusions. Am J Surg Pathol. 2019;43(10):1384–1391. doi:10.1097/PAS.0000000000001297
16. Kao Y, Suurmeijer AJH, Argani P, et al. Soft tissue tumors characterized by a wide spectrum of kinase fusions share a lipofibromatosis‐like neural tumor pattern. Genes Chromosomes Cancer. 2020;59(10):575–583. doi:10.1002/gcc.22877
17. Dermawan JK, Azzato EM, Goldblum JR, Rubin BP, Billings SD, Ko JS. Superficial ALK-rearranged myxoid spindle cell neoplasm: a cutaneous soft tissue tumor with distinctive morphology and immunophenotypic profile. Mod Pathol. 2021;34(9):1710–1718. doi:10.1038/s41379-021-00830-w

Esther Baranov, MD, is an instructor at Harvard Medical School and associate pathologist at Brigham and Women’s Hospital, Department of Pathology. She is board certified in Anatomic Pathology and Molecular Genetic Pathology. Dr. Baranov has research interests in bone and soft tissue, gastrointestinal, and molecular pathology.

Jason L. Hornick, MD, PhD, is the director of surgical pathology and immunohistochemistry and chief of soft tissue and bone pathology at Brigham and Women’s Hospital, professor of pathology at Harvard Medical School, and a consultant at the Dana-Farber Cancer Institute. Dr. Hornick’s research focuses on the development of diagnostic biomarkers for soft tissue tumors that correlate with molecular genetic alterations, which can be evaluated by conventional immunohistochemistry, and defining diagnostic criteria and clinicopathologic features of soft tissue tumors.