- Home
- Member Resources
- Articles
- DNA Methylation to the Rescue: An Update for Pathologists Navigating the Future of Precision Tumor Diagnostics
Introduction
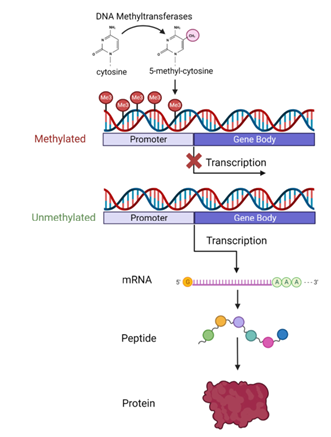
DNA methylation is an epigenetic modification whereby a methyl group is added to the cytosine nucleotide of DNA (Figure 1).1 This process plays a crucial role in regulating gene expression, often leading to gene silencing when occurring at promoter regions. In neoplastic processes, abnormal DNA methylation patterns, such as hypermethylation of tumor suppressor genes and/or global hypomethylation, contribute to tumorigenesis and tumor progression.

DNA methylation profiling assays are emerging technologies that are proving to be valuable assets in the field of pathology. By enabling more precise tumor classification, DNA methylation profiling can aid pathologists in challenging cases, enhancing diagnostic accuracy. Additionally, advancements in highly sensitive sequencing techniques now allow for cancer detection and preliminary classification through blood samples in some circumstances. To date, DNA methylation assays have shown promise in improving diagnosis, disease monitoring, and treatment decisions in a range of diverse tumor types.
This is exemplified in the latest World Health Organization (WHO) Classification of Tumors of the Central Nervous System, 5th edition, where the guidelines underscore the increasingly vital role of molecular diagnostics, particularly DNA methylation profiling, in the accurate classification of brain tumors. For medulloblastomas, the WHO specifically highlights the importance of combining molecular analysis, like DNA methylation profiling, with traditional morphological evaluation, noting that this integrated approach yields the most comprehensive prognostic information.2-4 Building on these advancements, this article explores the current applications, advancements, and future potential of DNA methylation profiling in improving tumor classification and diagnostics. It further emphasizes the technology's impact on precision medicine and the evolving role of pathologists in integrating and advancing these tools.
Figure 1: Diagram of DNA methylation1 A methyl group is added to cytosine (most often at cytosine-guanine dinucleotides) by DNA methyltransferase enzymes. Promoter region methylation is typically linked to gene silencing by decreasing transcription into mRNA.
Methodology and Limitations
The process of DNA methylation profiling typically begins with the extraction of DNA from tissue or blood samples, followed by bisulfite treatment, which converts unmethylated cytosines to uracil while leaving methylated cytosines unchanged. The sample is then analyzed using either array-based technologies, or sequencing methods, like whole-genome bisulfite sequencing (WGBS), for targeted or genome-wide assessment of methylation patterns, respectively. Bioinformatics tools are used to process the resulting data and compare them to reference databases, providing insights into tumor classification, origin, and potential therapeutic targets. Machine learning methods are employed to identify novel methylation signatures.
While a promising tool in cancer diagnostics, DNA methylation profiling faces several technical limitations. The cost and complexity of data, especially with WGBS, often necessitates significant computational resources and expertise, making it difficult to implement in routine clinical settings. Another limitation is the reliance on well-defined training datasets for accurate classification, meaning that incomplete reference data can lead to misclassification. Conversely, methylation classifiers can suffer from overfitting and may not generalize well to new, unseen data if the training set is not representative or comprehensive, as seen in rare tumor subtypes.5,6
Additionally, array-based methods, provide only partial coverage of the genome, potentially missing key methylation sites, whereas sequencing methods offer broader coverage but at a higher cost and with more complex bioinformatics demands. Variability in laboratory protocols and the need for specialized computational pipelines add further layers of operational complexity, preventing DNA methylation profiling from becoming a standardized diagnostic tool at the present time. Finally, a significant technical limitation is tumor purity in tissue samples, where low tumor cell content can limit the detection of tumor-specific DNA.6
For circulating tumor DNA (ctDNA) assays, a sufficient amount of ctDNA in the plasma is required for generating reliable results. Low ctDNA levels, often due to the tumor's location, small size, and/or reduced shedding, can lead to inconclusive or false-negative results. Many ctDNA technologies rely on a pipeline that must be trained with a robust set of cases to ensure proper calibration and result interpretation.7
Brain Tumors
For brain tumors, methylation-based classification has had a profound impact, particularly in refining the diagnosis of conditions like gliomas, medulloblastomas, and ependymomas. The development of the classifiers has allowed for rapid identification and differentiation of nearly all brain tumor types, providing precise tumor subgrouping where traditional histology was insufficient. For example, medulloblastomas are now categorized into four distinct molecular subgroups through methylation profiling, improving the clinical management of patients.5,8 Another example of DNA methylation classifiers improving brain tumor diagnosis is their use in distinguishing posterior fossa ependymomas. These tumors, previously classified based on histological features, are now divided into two molecular subgroups, Group A (more aggressive) and Group B, through DNA methylation profiling. This molecular classification better predicts clinical outcomes than the traditional histologic assessment of morphologic anaplasia.8
Studies have demonstrated that DNA methylation significantly improves tumor classification compared to histology alone. For example, one study revealed that using DNA methylation altered the initial diagnosis in 12% of CNS tumor cases.9 Additionally, in diagnostically challenging cases, where histological findings may be inconclusive or descriptive, DNA methylation has provided a definitive diagnosis in approximately 50% of cases.10,11
The neuropathologist is the crucial integrator of DNA methylation profiling with the more traditional workflows employed for brain tumor diagnosis and characterization. Histological analysis is not only vital for selecting the appropriate tissue for molecular testing but also for guiding the choice of molecular assays. For instance, a low-grade glioma may require BRAF fusion analysis, while a high-grade glioma would benefit from histone gene sequencing. Thoughtful selection and triage by the neuropathologist ensure that the most suitable diagnostic techniques are applied to each patient sample.12
Soft Tissue and Bone Tumors
DNA methylation analysis has shown potential as an aid in the diagnosis of sarcomas, providing a molecular approach to improve the accuracy of classification and diagnosis. Traditional histological evaluation of sarcomas can be challenging due to the overlapping features of different subtypes, making precise diagnosis difficult. DNA methylation profiling addresses these challenges, especially in ambiguous cases lacking distinct molecular alterations, by uncovering unique epigenetic signatures specific to various sarcoma types. One key advantage of this method is its ability to further subclassify undifferentiated sarcomas into more precise and reproducible subtypes, revealing a spectrum of tumors that would otherwise be grouped under the broad category of "not otherwise specified (NOS)."6,13,14
This approach extends to angiosarcoma, where the classifier can reveal molecular subgroups with significant clinical implications. In a large study on angiosarcoma, tumors were grouped into two main clusters, A and B, each with two subclusters, based on their methylation patterns. Cluster A was primarily composed of UV- or radiation-induced angiosarcomas and exhibited more chromosomal instability but better overall survival. In contrast, Cluster B included both primary and secondary angiosarcomas, with the majority of primary cases falling into this group. Interestingly, one subcluster within Cluster B (B1) contained seven cases with MGMT promoter methylation, indicating that this methylation status could potentially predict a response to treatments such as temozolomide.15
Another striking example of the use of the DNA methylation classifier is in diagnosing malignant peripheral nerve sheath tumors (MPNSTs). One study demonstrated that the classifier successfully identified MPNST in most cases. Remarkably, in 3 out of 18 cases, the classifier accurately revised the initial diagnosis to MPNST, a finding confirmed by loss of nuclear H3K27me3 expression. These included tumors originally classified as carcinosarcoma due to keratin-positive epithelioid components, dedifferentiated liposarcoma with MDM2 positivity and amplification, and a tumor resembling Ewing sarcoma.13 This underscores the potential pitfalls of relying solely on traditional diagnostic methods, as MPNSTs can present features like keratin positivity or MDM2 amplification.16 The ability of DNA methylation profiling to correctly identify MPNST in such scenarios underscores its use as a supplementary diagnostic tool in challenging cases.
Additionally, in the rapidly evolving soft tissue tumor classification area, both the potential utility and limitations of DNA methylation-based classifiers for novel tumor types has also been very recently illustrated. A newly characterized group of mesenchymal tumors harboring EGFR
kinase domain mutations was assessed using an array-based methylation platform and analyzed using an established methylation-based sarcoma classifier.17 This study found that none of the tumors achieved an optimal classification score and were assigned (incorrectly) to various entities. Yet, with less supervised analyses, the methylation profiles of these tumors clustered together in a well-defined group away from other entities, supporting the distinctness of this new tumor type and validity of methylation profiling as a technology to ascertain nosologically relevant differences between tumor types.
Other Tumors: Unknown primary carcinoma, hematolymphoid malignancies, and more
Epigenetic analysis also poses a valuable tool in diagnosing cancer of unknown primary. This approach helps by leveraging the unique methylation patterns that are specific to different tissue types and tumors. Since DNA methylation signatures are relatively stable and tissue-specific, profiling the methylome of a tumor can provide clues about its tissue of origin, even when histopathological features are inconclusive.2
In addition to its use in identifying the origin of unknown cancers, DNA methylation profiling is increasingly important in hematopathology, playing an important role in the diagnosis and risk stratification of conditions such as juvenile myelomonocytic leukemia (JMML). In JMML, distinct DNA methylation subgroups—low, intermediate, and high methylation—have been identified, each correlating with specific genetic mutations and clinical outcomes. For instance, the high methylation subgroup is closely associated with PTPN11 mutations, older age at diagnosis, and poorer overall survival, whereas the low methylation subgroup tends to be associated with more favorable outcomes. These methylation patterns have emerged as one of the strongest independent prognostic factors in JMML, providing a more precise method for patient stratification beyond traditional clinical and genetic markers.18 However, while early studies have shown encouraging results, integrating a methylation-based approach into the classification of hematopoietic tumors is likely to be more challenging compared to its application in CNS or soft tissue tumors, stemming from the fact that hematopoietic tumors already have a well-established and highly refined classification system based on specific genetic events.8
While DNA profiling is increasingly employed to assess prognosis in more common tumor types encountered in surgical pathology, DNA methylation analysis has demonstrated less diagnostic utility in kidney and thyroid tumors largely due to the relatively homogenous or less distinctive methylation patterns observed in these neoplasms. As a result, the additional insights from methylation profiling do not offer a substantial improvement in diagnostic accuracy for these tumors. Despite the lower diagnostic utility, the identification of distinct methylation subgroups in kidney and thyroid tumors may still play a valuable role in prognosis and therapy. These subgroups could reveal epigenetic alterations linked to specific biological pathways, potentially highlighting distinct tumor behavior or sensitivity to certain treatments, and leading to the identification of prognostically significant and therapeutically targetable subsets.6
ctDNA
Liquid biopsy technology has emerged as a critical tool in early cancer detection, particularly with the development of multi-cancer early detection (MCED) tests. These tests, which leverage both DNA and protein biomarkers, provide a non-invasive method to screen for various cancer types simultaneously. cfDNA methylation patterns alongside machine learning algorithms, has demonstrated significant potential in detecting cancer and predicting its tissue of origin.19 For instance, the PATHFINDER study demonstrated that MCED tests could identify a cancer signal in 1.4% of asymptomatic adults aged 50 and older, with 0.5% of participants confirmed to have cancer, highlighting the potential of these tests as a cancer screening tool.20
In colorectal cancer, the ECLIPSE trial has highlighted the utility of cfDNA-based screening tests, which achieved high sensitivity and specificity for advanced neoplasia. However, current cfDNA liquid biopsy tests still struggle with detecting precancerous polyps.21 Furthermore, several DNA methylation markers, including ADAMTS1, BNC1, and PXDN, have shown promise in early detection of pancreatic cancer and malignant risk stratification of intraductal papillary mucinous neoplasms.19
Future Directions and Conclusions
DNA methylation has emerged as a stable and reliable biomarker in cancer diagnostics, offering a robust reflection of the cell-of-origin for various tumors. Large-scale profiling has revealed that DNA methylation patterns are not only complex but also highly specific to particular tumor types. Moreover, DNA methylation as a biomarker is especially useful for histologically ambiguous tumors, as it can help arrive at a diagnosis. This technology, when combined with machine learning, has led to the identification of new tumor subtypes and the consolidation of histologically diverse tumors into more coherent groups.
Notably, the development of DNA methylation classifiers for CNS tumors and sarcomas represents a significant milestone in cancer diagnostics. These classifiers, which have already shown clinical utility, are likely to be followed by the development of similar tools for other cancer types. Beyond tissue-based classification, emerging methods are enabling the detection and classification of cancer from blood samples, pointing toward a future where liquid biopsies may become useful for early detection, preliminary diagnosis, and monitoring in routine care.
While once a distant possibility, the advancement of DNA methylation techniques is now bringing the potential for a pan-cancer DNA methylation classifiers closer to realization. The utility of such a classifier lies in its ability to confirm and enhance histomorphology-based diagnoses across the broad spectrum of different pathology subspecialties, identify clinically relevant subtypes, serve as a quality control and educational resource, and play a pivotal role in discovering new neoplastic entities. The development of these classifiers will rely heavily on pathologists, who must play a critical role in their creation and implementation. In this future, pathologists will likely integrate methylation profiling data, alongside other molecular findings, with traditional diagnostic methods for many types of cases, especially those that are diagnostically challenging. The result will be more precise and comprehensive, significantly enhancing patient care and improving outcomes across a wide range of cancers.6,22
References
- Wang SS, Lewis MJ, Pitzalis C. DNA Methylation Signatures of Response to Conventional Synthetic and Biologic Disease-Modifying Antirheumatic Drugs (DMARDs) in Rheumatoid Arthritis. Biomedicines. Jul 13 2023;11(7) doi:10.3390/biomedicines11071987. Reprinted under Creative Commons license CC-BY-4.0
- Davalos V, Esteller M. Cancer epigenetics in clinical practice. CA Cancer J Clin. Jul-Aug 2023;73(4):376-424. doi:10.3322/caac.21765
- Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. Aug 2 2021;23(8):1231-1251. doi:10.1093/neuonc/noab106
- WHO Classification of Tumours Editorial Board. Central Nervous System Tumours. WHO Classification of Tumours, 5th Edition. IARC Press; 2021.
- Sill M, Plass C, Pfister SM, Lipka DB. Molecular tumor classification using DNA methylome analysis. Hum Mol Genet. Oct 20 2020;29(R2):R205-R213. doi:10.1093/hmg/ddaa147
- Papanicolau-Sengos A, Aldape K. DNA Methylation Profiling: An Emerging Paradigm for Cancer Diagnosis. Annu Rev Pathol. Jan 24 2022;17:295-321. doi:10.1146/annurev-pathol-042220-022304
- Pascual J, Attard G, Bidard FC, et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: a report from the ESMO Precision Medicine Working Group. Ann Oncol. Aug 2022;33(8):750-768. doi:10.1016/j.annonc.2022.05.520
- Koelsche C, von Deimling A. Methylation classifiers: Brain tumors, sarcomas, and what's next. Genes Chromosomes Cancer. Jun 2022;61(6):346-355. doi:10.1002/gcc.23041
- Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. Aug 2018;136(2):181-210. doi:10.1007/s00401-018-1879-y
- Galbraith K, Snuderl M. DNA methylation as a diagnostic tool. Acta Neuropathol Commun. May 8 2022;10(1):71. doi:10.1186/s40478-022-01371-2
- Wu Z, Abdullaev Z, Pratt D, et al. Impact of the methylation classifier and ancillary methods on CNS tumor diagnostics. Neuro Oncol. Apr 1 2022;24(4):571-581. doi:10.1093/neuonc/noab227
- Cotter JA, Judkins AR. Fitting the epigenome into the picture: methylation classification for paediatric brain tumours. Neuropathol Appl Neurobiol. Oct 2018;44(6):543-547. doi:10.1111/nan.12488
- Miettinen M, Abdullaev Z, Turakulov R, et al. Assessment of The Utility of The Sarcoma DNA Methylation Classifier In Surgical Pathology. Am J Surg Pathol. Jan 1 2024;48(1):112-122. doi:10.1097/PAS.0000000000002138
- Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. Jan 21 2021;12(1):498. doi:10.1038/s41467-020-20603-4
- Weidema ME, van de Geer E, Koelsche C, et al. DNA Methylation Profiling Identifies Distinct Clusters in Angiosarcomas. Clin Cancer Res. Jan 1 2020;26(1):93-100. doi:10.1158/1078-0432.CCR-19-2180
- Makise N, Sekimizu M, Kubo T, et al. Clarifying the Distinction Between Malignant Peripheral Nerve Sheath Tumor and Dedifferentiated Liposarcoma: A Critical Reappraisal of the Diagnostic Utility of MDM2 and H3K27me3 Status. Am J Surg Pathol. May 2018;42(5):656-664. doi:10.1097/PAS.0000000000001014
- Vallese S, Barresi S, Hiemcke-Jiwa L, et al. Spindle Cell Lesions with Oncogenic EGFR Kinase Domain Aberrations: Expanding the Spectrum of Protein Kinase-Related Mesenchymal Tumors. Mod Pathol. Sep 2024;37(9):100539. doi:10.1016/j.modpat.2024.100539
- Schonung M, Meyer J, Nollke P, et al. International Consensus Definition of DNA Methylation Subgroups in Juvenile Myelomonocytic Leukemia. Clin Cancer Res. Jan 1 2021;27(1):158-168. doi:10.1158/1078-0432.CCR-20-3184
- Tan WY, Nagabhyrava S, Ang-Olson O, et al. Translation of Epigenetics in Cell-Free DNA Liquid Biopsy Technology and Precision Oncology. Curr Issues Mol Biol. Jun 27 2024;46(7):6533-6565. doi:10.3390/cimb46070390
- Schrag D, Beer TM, McDonnell CH, 3rd, et al. Blood-based tests for multicancer early detection (PATHFINDER): a prospective cohort study. Lancet. Oct 7 2023;402(10409):1251-1260. doi:10.1016/S0140-6736(23)01700-2
- Chung DC, Gray DM, 2nd, Singh H, et al. A Cell-free DNA Blood-Based Test for Colorectal Cancer Screening. N Engl J Med. Mar 14 2024;390(11):973-983. doi:10.1056/NEJMoa2304714
- Nakhleh RE, Nose V, Colasacco C, et al. Interpretive Diagnostic Error Reduction in Surgical Pathology and Cytology: Guideline From the College of American Pathologists Pathology and Laboratory Quality Center and the Association of Directors of Anatomic and Surgical Pathology. Arch Pathol Lab Med. Jan 2016;140(1):29-40. doi:10.5858/arpa.2014-0511-SA

Matthew Hiemenz, MD, MS, FCAP, is a senior pathologist and associate medical director at Foundation Medicine. He is board-certified in anatomic and clinical pathology, molecular genetic pathology, and clinical informatics. His interests include assay validation and evidence generation for new assays and biomarkers, biomarker-driven trial design, and the molecular pathology of pediatric solid tumors. He serves on the College of American Pathologists Personalized Health Care Committee.

Justin Chang, MD, is a fourth-year AP/CP resident a Virginia Commonwealth University Health System and a junior member on the College of American Pathologists Digital and Computational Pathology Committee.