- Home
- Member Resources
- Articles
- Biomarker Testing in Gastrointestinal Stromal Tumors (GIST)
Gastrointestinal stromal tumors (GIST) are the most common type of mesenchymal tumors of the digestive tract. GIST may occur anywhere in the GI tract, but stomach [60% of cases] and small intestine [30%] are the most common sites. From molecular genetics points of view, GIST can be grouped into 4 major molecularly-defined groups: 1) KIT mutated [about 75 to 80%], 2) PDGFRA mutated [about 5 to 10%], 3) SDH-deficient [about 5 to 10%], and 4) other rare driver mutations [NF1, NTRK, BRAF etc].1-11
The KIT gene encodes the stem cell factor receptor and PDGFRA gene encodes the platelet-derived growth factor receptor alpha; both belong to the type III tyrosine kinase receptor family. The mutations in KIT and PDGFRA lead to constitutive receptor tyrosine kinase activity. KIT and PDGFRA mutations are almost always mutually exclusive, but activate similar downstream pathways, including the JAK–STAT3, phosphatidylinositide-3-kinase (PI3K)–AKT–mTOR, and RAS–MAPK pathways, which are all important in regulating proliferation and apoptosis leading to oncogenesis.1-7
KIT Mutations

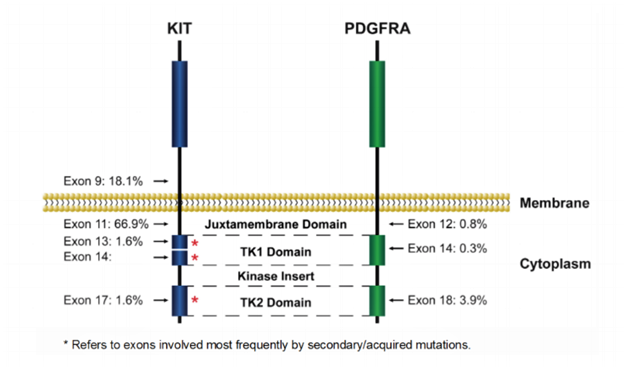
For cases involving KIT, mutations cluster within “hot spots”: exons 9, 11, 13, 17 (Figure 1 and Table 1).1-8
Exon 11, encoding the juxtamembrane domain, is the most frequently mutated region, affecting 60–70% of GIST. The mutations disrupt the autoinhibitory domain of the receptor and permit continuous kinase activation. Deletions affecting codons 557-558 of exon 11 (Trp557_Lys558del) are the most common, detected in 23-28% of GISTs. Point mutations, duplications and homo/hemizygous mutation have also been identified. The vast majority (>80%) of KIT exon 11-mutated GISTs are in the stomach, although they may occur anywhere in the GI tract.
Mutations in KIT exon 9, encoding part of the extracellular domain, occur in 10–20% of GIST cases (Table 1). 1-8 These mutations are believed to result in dimerization of KIT leading to constitutive activation. The majority of exon 9 mutations are duplications of codons 502 and 503 and are almost exclusively associated with GISTs in small intestine. Mutations in exons 13 and 17 are uncommon, occurring in 1-2% of GISTs. Mutations in exon 17 encoding the activation loop of the kinase may stabilize the active conformation and occur more frequently in the small intestinal GISTs.
GIST Biomarker Summary (Table 1)
The current NCCN guideline recommends KIT and PDGFRA mutational analysis for patients needing tyrosine kinase inhibitor (TKI) treatment for unresectable or metastatic disease and for patients with high-risk tumors determined by clinical and pathological features.7 Most patients with KIT mutations respond well to imatinib at a dose of 400 mg daily when TKI treatment is indicated, with exception of KIT exon 9 mutated tumors for which the 400 mg twice daily dose can be considered. Of note, sunitinib has been reported to induce higher response rates, longer progression free survival (PFS) and overall survival (OS) for patients with KIT exon 9 mutations compared to patients with KIT exon 11 mutations. Of note, although KIT expression via IHC (CD117) may be helpful as a diagnostic marker of GIST, it is unrelated to underlying KIT mutations and is no longer considered adequate for predicting response to anti-KIT therapies.
Primary imatinib resistance, defined as progressive disease developing within the first 6 months of treatment, is most seen in patients with KIT exon 9 mutations with the standard dose of imatinib (400mg/day).7 Secondary imatinib resistance is defined as patients on imatinib for more than 6 months with an initial response or stable disease followed by disease progression. Secondary resistance is most associated with cancer cells developing secondary mutations in KIT. Secondary mutations are usually in exons 13, 14, and 17. Patients with secondary KIT exon 13 or 14 mutations usually respond to sunitinib while secondary exon 17 or 18 mutations usually respond to regorafenib or ripretinib.1-2,7-8
PDGFRA Mutation
PDGFRA-mutated GISTs are mostly of gastric origin and display epithelioid cell morphology or mixed epithelioid and spindle cell histology. PDGFRA mutations occur mainly in exons 12 and 18 and rarely in exon 14 (Figure 1 and Table 1).1-2,7-10 PDGFRA mutations affecting exon 18 (the second kinase domain), likely resulting in stabilizing the kinase activation loop, have been identified in approximately 4 to 6% of GISTs. The most frequent exon 18 mutation is p.D842V. Mutations affecting exon 12 (the juxtamembrane domain) are the second most common type of PDGFRA mutation and occur in about 1–2% of GISTs. The intact PDGFRA juxtamembrane domain may have an autoinhibitory function and mutations lead to hyperactivation. Mutations in exon 14 frequently cluster at codon 659 and are rare (<0.1% of GISTs).
The most common mutation in PDGFRA, p.D842V, has been shown to be associated with primary resistance to imatinib and most TKIs except avapritinib when systemic treatment is indicated.1-2,7-8 Avapritinib was recently approved by FDA to treat GIST cases with PDGFRA exon 18 mutations (January 2020). PDGFRA exon 12 mutations and other PDGFRA mutations are generally sensitive to imatinib.
SDH-deficient GISTs
Approximately 10 to 15% of GIST cases lack KIT or PDGFRA mutations. Most of these cases have inactivation of the succinate dehydrogenase (SDH) complex ("SDH-deficient GISTs").1-2, 11 SDH deficiency can be readily detected by the lack of SDHB expression using immunohistochemical staining which should be performed on all GIST cases lacking KIT or PDGFR mutation. SDH is a mitochondrial enzyme complex with four subunits—SDHA, SDHB, SDHC, and SDHD. Deficiency of SDH complex leads to accumulation of succinate and may result in hypoxia-inducible factor (HIF)1-α stabilization and constitutive activation of hypoxic signaling and tumorigenesis.
Inactivation of SDH complex may result from mutations of SDH complex subunit genes (SDHA/B/C or D) or epigenetic silencing. Around 50% of SDH-deficient GIST patients harbor germline mutations in one of the SDH subunit genes (Table 1).1-2, 11 SDHA-inactivating mutations are most common, detected in about 30% of SDH-deficient GISTs. If no mutation of SDH complex subunit genes is identified, SDHC promoter methylation should be evaluated.1-2, 11
GIST with SDH complex mutations usually occur in the stomach of young individuals (frequently female), often metastasize to liver and peritoneum, may involve lymph nodes and typically grow slowly with an indolent long-term clinical course. These tumors show multilobulated/multinodular growth pattern with epithelioid morphology.
SDH-deficient GIST frequently present as either Carney- Stratakis (CS) syndrome or Carney triad1-2, 11. CS syndrome, caused by germline mutations of SDH complex subunit genes, is a hereditary predisposition syndrome characterized by multiple gastric GISTs and paragangliomas. Carney-triadis characterized by multiple gastric GISTs, paragangliomas, and pulmonary chondromas. Carney-triad is usually associated with epigenetic SDH inactivation often via SDHC hypermethylation but has been rarely associated with germline mutations of SDH complex subunit genes.
SDH-deficient GIST are usually resistant to imatinib but may benefit from therapy with other TKIs (such as sunitinib) when systemic treatment is needed.7
Genetic counselling is indicated for patients with germline mutations of SDH subunit genes. Additionally, regular screening for paraganglioma, pheochromocytoma, and SDH-deficient renal cell carcinoma and pituitary tumors is recommended for these patients.
Rare Mutations
For cases that are “triple-negative”, testing for alternative driver variants (e.g., BRAF mutation, NF1 mutation, NTRK fusions) can be done for potential targeted therapies. GIST associated with neurofibromatosis type 1 (NF1), an autosomal dominant disease, predominantly occur in the small intestine and can be multicentric. Genetic counselling is indicated for these patients.
In summary, most GIST cases are associated with known molecular genetics pathways. GIST patients usually respond well to targeted therapies based on the driver mutations discussed above. For patients who are considered for PD1/PD-L1 blockade treatment, tumor mutation burden and microsatellite instability studies are recommended.
References:
- Blay JY, Kang YK, Nishida T, von Mehren M. Gastrointestinal stromal tumours. Nat Rev Dis Primers. 2021;7(1):1-22.
- Parab TM, DeRogatis mj, Boaz AM, Gastrointestinal stromal tumors: a comprehensive review, J Gastrointest Oncol. 2019;10:144–154
- Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21(23):4342-4349. https://ascopubs.org/journal/jco
- Verweiji J, Casali PG, Zaleberg J, et al. Progression–free survival in gastrointestinal stromal tumors with high-dose imatinib: randomized trial. Lancet. 2004;364(9440):1127-1134.
- Falchook GS, Trent JC, Heinrich MC, et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4:310-5
- My Cancer Genome https://www.mycancergenome.org/content/biomarkers; accessed 5/11/202NCCN Guideline, Gastrointestinal stromal tumors, version 1.2021
- NCCN Guideline, Gastrointestinal stromal tumors, version 1.2021
- OncoKB https://www.oncokb.org; accessed 5/11/2022
- https://oncologypro.esmo.org/education-library/factsheets-on-biomarkers/pdgfra-in-gastrointestinal-stromal-tumours-gist; last accessed 5/11/2022
- https://oncologypro.esmo.org/education-library/factsheets-on-biomarkers/kit-and-pdgfra-wild-type-gastrointestinal-stromal-tumours-gists; last accessed 5/11/2022
- https://oncologypro.esmo.org/education-library/factsheets-on-biomarkers/kit-in-gastrointestinal-stromal-tumours-gist; last accessed 5/11/2022

Chung-Che (Jeff) Chang, MD, PhD, FCAP, is the medical director of Hematology and Molecular/Genomic Laboratory at AdventHealth-Orlando, and professor of pathology, University of Central Florida. He currently serves as associate editor for Archives of Pathology and Laboratory Medicine, and a member of the Personalized Healthcare Committee for the College of American Pathologists (CAP). He was the principal investigator of several National Institutes of Health (NIH)/National Cancer Institute (NCI) grants to study myeloma, myelodysplastic syndromes, and lymphoma. Dr. Chang’s research interests include the use of next-generation sequencing technologies for clinical diagnostics and biomarker discovery.